|
|
|
|
|
|
 Hauptseminar im WS1999/2000:
Hauptseminar im WS1999/2000:Das Hauptseminar findet donnerstags von 14:15 Uhr bis 15:45 Uhr im Raum S2229 statt.
| 16.12.1999: |
Alex Souza Protein Folding: HP-Model and Approximations Betreuer: Volker Heun |
| 13.01.2000: |
Florian Rodler Sequence Alignment: Dynamic Programming Betreuer: Jens Ernst |
| 20.01.2000: |
Sueleyman Aydogmus Sequence Alignment: Suffix Trees and Common Substrings Betreuer: Jens Ernst |
| 10.02.2000: |
Christoph Bodensteiner. Evolutionary Trees: Construction from Additive matrices Betreuer: Volker Heun |
| 17.02.2000: |
Anton Epple Protein Folding: Inverse Protein Folding Betreuer: Volker Heun |
| 24.02.2000: |
Andreas Kiermeier Genome Rearrangements: Sorting Signed Permutations by Reversals and Transpositions Betreuer: Jens Ernst |
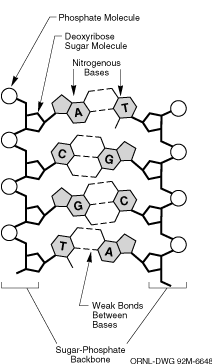 Das Sequence Alignment ist eines der fundamentalsten und ältesten
Probleme der Bioinformatik.
Hierbei soll für zwei gegebenen Zeichenreihen (interpretiert als
Abfolge von Nuklein- oder Aminosäuren) festgestellt werden, ob
sie ähnlich sind, und wenn ja, wie sie mit einer minimalen Anzahl
elementarer Operationen (Löschen, Einfügen, Substitution von
Zeichen) ineinander überführt werden können.
Werden zum Beispiel mehrere gleichen DNS-Sequenzen bzw. Proteine
sequenziert, so will man hiermit feststellen, ob bzw. inwieweit die
Ergebnisse dieselben sind.
Ein anderes Beispiel (in dem man eine Sequenz mit vielen anderen
vergleicht) ist die Suche in einer DNS- bzw. Protein-Datenbank, wobei
man feststellen möchte, ob die neue Sequenz schon bekannt ist
oder nicht.
Das Sequence Alignment ist eines der fundamentalsten und ältesten
Probleme der Bioinformatik.
Hierbei soll für zwei gegebenen Zeichenreihen (interpretiert als
Abfolge von Nuklein- oder Aminosäuren) festgestellt werden, ob
sie ähnlich sind, und wenn ja, wie sie mit einer minimalen Anzahl
elementarer Operationen (Löschen, Einfügen, Substitution von
Zeichen) ineinander überführt werden können.
Werden zum Beispiel mehrere gleichen DNS-Sequenzen bzw. Proteine
sequenziert, so will man hiermit feststellen, ob bzw. inwieweit die
Ergebnisse dieselben sind.
Ein anderes Beispiel (in dem man eine Sequenz mit vielen anderen
vergleicht) ist die Suche in einer DNS- bzw. Protein-Datenbank, wobei
man feststellen möchte, ob die neue Sequenz schon bekannt ist
oder nicht.
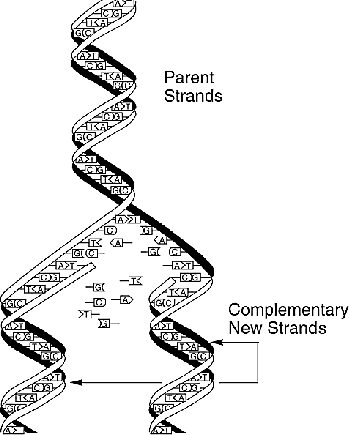 Aus biotechnologischen Gründen können immer nur relativ
kurze Stücke eines DNS-Strangs sequenziert werden.
Für eine vollständige Strukturauflösung eines langen
DNS-Strangs muß dieser also in viele kurze aufgeteilt werden,
wobei in Wirklichkeit die Bruchstücke vieler identischer Kopien
dieses DNS-Strangs betrachtet werden.
Beim Fragment Assembly will man nun aus dieses Teilsequenzen wieder
die gesamte Sequenz des langen DNS-Strangs rekonstruieren.
Aus biotechnologischen Gründen können immer nur relativ
kurze Stücke eines DNS-Strangs sequenziert werden.
Für eine vollständige Strukturauflösung eines langen
DNS-Strangs muß dieser also in viele kurze aufgeteilt werden,
wobei in Wirklichkeit die Bruchstücke vieler identischer Kopien
dieses DNS-Strangs betrachtet werden.
Beim Fragment Assembly will man nun aus dieses Teilsequenzen wieder
die gesamte Sequenz des langen DNS-Strangs rekonstruieren.
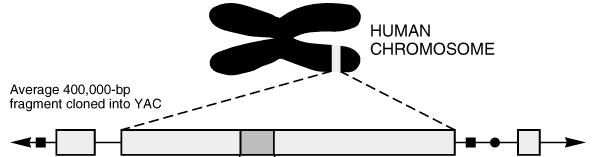 Hat man Bereiche von DNS-Strängen eines Chromosoms sequenziert,
so will man natürlich auch wissen, wo in diesem Chromosom ein
sequenzierter Teil vorkommt.
Biotechnisch können dabei bestimmte sehr kurze Teile des
Chromosoms markiert werden.
Diese Markierungen kann man auf den Bruchstücken des DNS-Strangs
wiederfinden.
Ziel ist es nun, die Bruchstücke so zusammenzusetzen, daß
die markierten Stellen auf dieser Zusammensetzung an denselben Stellen
sind wie im ursprünglichen Chromosom.
Hat man Bereiche von DNS-Strängen eines Chromosoms sequenziert,
so will man natürlich auch wissen, wo in diesem Chromosom ein
sequenzierter Teil vorkommt.
Biotechnisch können dabei bestimmte sehr kurze Teile des
Chromosoms markiert werden.
Diese Markierungen kann man auf den Bruchstücken des DNS-Strangs
wiederfinden.
Ziel ist es nun, die Bruchstücke so zusammenzusetzen, daß
die markierten Stellen auf dieser Zusammensetzung an denselben Stellen
sind wie im ursprünglichen Chromosom.
 Vergleicht man das gesamte Genom von verschiedenen Spezies, so
hätte man gerne einen Abstandsbegriff, der für ein Paar von
Spezies aussagt, wie verwandt sie miteinander sind.
Bei vielen Spezies unterscheiden sich das Erbgut oft nur durch eine
andere Anordnung der Gene innerhalb des Erbguts.
Daher definiert man den Abstandsbegriff durch die minimale Anzahl an
gewissen Elementaroperationen, die nötig sind, um die Genome der
Spezies ineinander zu überführen.
Dabei sind hier die elementaren Operationen (im Gegensatz zum Sequence
Alignment) globale Operationen.
Am häufigsten treten sogenannte Inversionen und Transpositionen
auf, bei denen entweder ein ganzer Abschnitt der DNS-Strangs umgedreht
wird oder zwei lange benachbarte Abschnitte miteinander vertauscht
werden.
Vergleicht man das gesamte Genom von verschiedenen Spezies, so
hätte man gerne einen Abstandsbegriff, der für ein Paar von
Spezies aussagt, wie verwandt sie miteinander sind.
Bei vielen Spezies unterscheiden sich das Erbgut oft nur durch eine
andere Anordnung der Gene innerhalb des Erbguts.
Daher definiert man den Abstandsbegriff durch die minimale Anzahl an
gewissen Elementaroperationen, die nötig sind, um die Genome der
Spezies ineinander zu überführen.
Dabei sind hier die elementaren Operationen (im Gegensatz zum Sequence
Alignment) globale Operationen.
Am häufigsten treten sogenannte Inversionen und Transpositionen
auf, bei denen entweder ein ganzer Abschnitt der DNS-Strangs umgedreht
wird oder zwei lange benachbarte Abschnitte miteinander vertauscht
werden.
 Die Funktionalität eines Proteins wird im wesentlichen durch
seine räumliche Struktur bestimmt.
Leider sind physikalische Strukturaufklärungsverfahren im
Vergleich zur biologischen Sequenzierung sehr aufwendig.
Deshalb ist man an Algorithmen interessiert, die aus der
Aminosäuresequenz eines Proteins die räumliche Struktur des
Proteins vorhersagen oder die zu einer gegebenen räumlichen
Struktur eine geeignete Aminosäuresequenz angeben, die sich in
die vorgegebene Struktur faltet.
Die Funktionalität eines Proteins wird im wesentlichen durch
seine räumliche Struktur bestimmt.
Leider sind physikalische Strukturaufklärungsverfahren im
Vergleich zur biologischen Sequenzierung sehr aufwendig.
Deshalb ist man an Algorithmen interessiert, die aus der
Aminosäuresequenz eines Proteins die räumliche Struktur des
Proteins vorhersagen oder die zu einer gegebenen räumlichen
Struktur eine geeignete Aminosäuresequenz angeben, die sich in
die vorgegebene Struktur faltet.
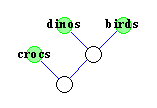 Durch die Evolution haben sich die verschiedenen Spezies aus
gemeinsamen Vorfahren weiterentwickelt, die zum Teil schon lange
wieder ausgestorben sind.
Mit Hilfe eines phylogenetischen Baumes will man nun den Stammbaum
verschiedener Spezies rekonstruieren, wobei man nur die noch lebenden
Spezies kennt.
Diese lebenden Spezies bilden nun die Blätter des
phylogenetischen Baumes.
Durch die Evolution haben sich die verschiedenen Spezies aus
gemeinsamen Vorfahren weiterentwickelt, die zum Teil schon lange
wieder ausgestorben sind.
Mit Hilfe eines phylogenetischen Baumes will man nun den Stammbaum
verschiedener Spezies rekonstruieren, wobei man nur die noch lebenden
Spezies kennt.
Diese lebenden Spezies bilden nun die Blätter des
phylogenetischen Baumes.
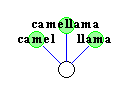 Ziel ist nun, die inneren Knoten des Baumes so zu konstruieren,
daß stark verwandte Spezies im Baum nah zusammen (durch einen
kürzeren Pfad verbunden) sind.
Zur Konstruktion phylogenetischer Bäume werden dabei
unterschiedliche Methoden zur Bestimmung des Verwandtschaftsgrades
verwendet.
Ziel ist nun, die inneren Knoten des Baumes so zu konstruieren,
daß stark verwandte Spezies im Baum nah zusammen (durch einen
kürzeren Pfad verbunden) sind.
Zur Konstruktion phylogenetischer Bäume werden dabei
unterschiedliche Methoden zur Bestimmung des Verwandtschaftsgrades
verwendet.
Die detaillierte Themenliste mit Literaturangaben finden Sie hier.
|
|
Merkblatt zur Gestaltung eines Seminarvortrags. (Die Tips auf diesem Merkblatt sind keine offiziellen Anforderungen oder Bewertungskriterien der TU München, sondern aus der Praxis eines Seminarleiters heraus entstandene Ratschläge.) |
|
|
Tips zur Erstellung von Folien mit LaTeX (einschließlich Rahmen-Datei als Vorlage) |
Weitere Auskünfte erteilen Jens Ernst und Volker Heun.
Volker Heun, 1999-08-25